
Abstract
Gene Regulatory Networks (GRNs) inference from gene expression data is a hard task and a widely addressed challenge. GRNs can be represented as Boolean models similarly to digital circuits. Cartesian Genetic Programming (CGP), often used for designing circuits, can thus be adopted in the inference of GRNs. The main CGP operator for generating candidate designs is mutation, making its choice important for obtaining good results. Although there are many mutation operators for CGP, to the best of our knowledge, there is no analysis of them in the GRN inference problem. An evaluation of the Single Active Mutation (SAM) and the Semantically-Oriented Mutation Operator (SOMO) is performed here for GRNs inference. Also, a combination of both operators is proposed. We use a benchmark single-cell RNA-Sequencing time series data and its evaluation pipeline to measure the performance of the approaches. The experiments indicate that (i) combining SOMO and SAM provides the best results, and (ii) the results obtained by the proposal are competitive with those from state-of-the-art methods.
Access provided by University of Notre Dame Hesburgh Library. Download conference paper PDF
Similar content being viewed by others

1 Introduction
Systems Biology is an interdisciplinary research area that focuses on the computational and mathematical analysis of interactions among the components of a biological system [28]. All cellular activities are controlled by their genes through a network that forms proteins from DNA. The gene expression depends on the relationships in this network, known as Gene Regulatory Network (GRN) [13].
Several methods have been developed for the inference of GRNs, such as PIDC [3], that uses partial information decomposition (PID) to identify regulatory relationships between genes, GENIE3 [12], winner of the DREAM5 network challenge, that decomposes the prediction of a regulatory network among p genes into p different regression problems, and GRNBoost2 [19], that is based on GENIE3, and uses a Gradient Boosting Machine (GBM) regression [7]. GENIE3 and GRNBoost2 are provided in the same Python package Arboreto [19]. Also, evolutionary computation techniques have been applied to infer gene regulatory networks. For instance, one can find the application of Genetic Programming and Evolutionary Strategies in this context [15, 26].
GRNs can be modeled as continuous models in the form of differential equations or as discrete models, such as Bayesian and Boolean networks. For discrete models, a common scheme for representing the genes’ interactions is an alphabet of two symbols \(\Gamma \) = {0,1}, where 0 means inhibition and 1 means activation. A model for this type of network is similar to digital circuits, which can be evolved using Cartesian Genetic Programming (CGP) [18], as presented in [24, 25].
Although it is possible to use recombination operators, CGP mostly uses mutation operators to create genetic variation in its offspring. There is no general-purpose crossover operator with good performance for CGP [17]. Point-mutation, in which a gene is randomly chosen and changed to another value, and Single Active Mutation (SAM) [9] that guarantees a phenotypic difference after the mutation, are two common mutation approaches used on CGP.
Recently, the Semantically-Oriented Mutation Operator (SOMO) was developed for designing evolutionary circuits [11]. SOMO can obtain feasible circuits using less computational effort than other approaches from the literature, and this advantage helps to deal with the scalability limitation of CGP. However, SOMO uses only a specific population initialization in order to optimize the circuit in terms of reducing the number of logic elements. There is not a proper optimization step. Also, point-mutation has a parameter, \(\mu _{r}\), which defines the percentage of the genotype to be mutated. SOMO, on the other hand, has more sensitive parameters that control the genetic variation of the offspring.
Therefore, in this paper, we propose the evaluation and analysis of CGP when applied to the context of the gene regulation networks inference by using benchmark curated models with single-cell RNA-Sequencing (scRNA-Seq) time-series gene expression data technology. Here, we provide a critical assessment of the advantages and disadvantages of using SOMO, with and without an appropriate optimization scheme with SAM. Furthermore, we perform a parameter sensitivity analysis of SOMO. Also, the results obtained by the proposal in the computational experiments are better than those found by other GP approaches and similar to those reached by the state-of-the-art algorithm GENIE3, one of the techniques more widely used in the literature for modeling GRNs. Our results show that it is important to have an optimization step to obtain correct GRNs and that SOMO is very sensitive to its parameters.
2 Problem Definition
The usual research aim is to infer the network topology from given gene expression data [1]. The gene expression data can be given as a steady-state or time-series matrix. For the former, each measure represents the expression level of a given gene in a certain condition and, for the latter, the gene expression measurements is carried out in each time point. Considering N the total number of genes and S the total number of time-points measured, a gene expression dataset is represented by a S x N matrix, where each row vector s (s = 1, ..., S) represents a N-dimensional transcriptome, and each column vector y (y = 1, ..., N) corresponds to a S-dimensional gene profile in the total cell population [4]. The goal of the network inference method is to use the data matrix to predict a set of regulatory interactions between any two genes from the total of N genes. The final output is in the form of a graph with N nodes and a set of edges [4].
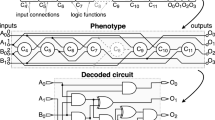
Figure 1a shows an example of a GRN with four genes (nodes) and their regulatory relationships (edges). Blue pointed arrows represent an activation and orange lines, inhibition. For example, G1 activates G2, and G4 inhibits G3.

Illustrations of a GRN with four genes (G1-G4) (a) and an individual of CGP (b). The representation is composed by three primary inputs (I0, I1, and I2) and one output (O0). Continuous lines defines the phenotype. In this representation, \(n_{c}\) = \(n_{r}\) = 3 and the function set \(\Gamma \) = {AND, OR, NOT, XOR}.
Discrete and continuous models of GRNs are often used to understand the process. When considering continuous models, it is common to use differential equations to model the regulatory relationships between genes, such as presented in [8, 24]. This type of model uses the concentrations of the macromolecules such as RNAs and proteins (both are gene products) and other biological species concentration, modeling them through the time-rate-of-change of their concentration variables. Regulatory interactions take the form of functional and differential relationships between concentration variables [22]. More specifically, gene regulation is modeled by rate equations that express the rate of production of a system component as a function of the concentrations of other components.
Also, it is possible to model GRNs through a discrete model, such as Boolean Networks. Boolean Networks use Boolean Algebra to discover the relationship between genes. Moreover, Boolean networks assume each gene g at a time point t to be in one of two states, active or inactive, according to its gene expression data at time t [2], therefore, one needs to binarize the data, leading to information loss. Given a threshold, the data is discretized in 1 (activation) or 0 (inhibition). Boolean-based models simplify the structure and dynamics of gene regulation. Inferred networks provide a quantitative measure of gene regulatory mechanisms [22]. This model, despite its simplicity, can represent, through its dynamics, several biologically significant phenomena. Furthermore, it is possible to obtain several practical uses, such as the identification of drugs for cancer treatment through the inference of the relationships between genes from experimental data such as gene expression profiles [16].
There are several techniques for measuring the expression of a gene. For instance, single-cell RNA-Sequencing (scRNA-seq) is attractive for GRN inference due to its production of thousands of independent measurements [14] and there are methods that sort cells along “trajectories” describing the development or progress of the cell [10, 21, 23, 27], called pseudotime, which is a measure of how far a cell has moved through biological progress. Also, when using scRNA-Seq data technologies, it is common to observe a dropout, which occurs when a gene is observed at a low or moderate expression level in one cell but it is not detected in another cell of the same type. Dropouts make it difficult to get correct GRNs.
3 Cartesian Genetic Programming
Cartesian Genetic Programming is a Genetic Programming technique in which programs are Directed Acyclic Graphs (DAGs) encoded by a matrix of processing nodes, with \(n_{c}\) columns and \(n_{r}\) rows. [17]. The genes are integer values and, for each gene, there are inputs and operation/function that the node performs.
Given a node in the matrix, the nodes at its left side can be used as inputs. There is a user-defined parameter (levels-back) that limits the number of columns at the left side where inputs can be selected to constrain the connectivity of the graph. The number of inputs that a node can receive is called arity. Also, the genotype contains nodes that contribute directly to the output, called active nodes, and those that do not, the inactive nodes. The phenotype is composed only of the active nodes and the genotype-phenotype mapping is done by recursively determining the nodes that contribute to each output, starting on the output and ending on the primary inputs. The function set is user-defined and problem-dependent. For example, logic functions or gates are used when designing digital circuits. Figure 1b presents a CGP individual with three primary inputs and one output. The functions are logical ones. Grey nodes are active and white nodes, inactive. The most common search technique used in CGP is the \((1 + \lambda )\) Evolutionary Strategy (ES) [17], where \(\lambda \) is the number of new solutions generated at each iteration. In this case, the best individual, the one with more matches concerning its truth-table, between the parent and the \(\lambda \) new generated solutions is selected for the next generation.
3.1 Mutation Operators
CGP normally uses only mutation to generate new individuals and crossover operators have received little attention in the CGP [17]. The two most adopted mutation approaches are point mutation [17] and SAM [9].
Point mutation is a simple approach that randomly selects a node and a gene and changes it to another valid value. However, the changes may occur in inactive nodes leading to a lack of modifications in the phenotype.
SAM, proposed for reducing the number of wasted objective function evaluations, operates by (i) randomly selecting one node and one of its elements (inputs or function) and (ii) changing its value to another valid value. Steps (i) and (ii) are repeated until an active node is modified. As a result, SAM ensures that one active node is changed.
SOMO [11] was developed for the design of digital circuits using CGP, where the purely stochastic mutation operator is replaced and operates in the phenotype space. The SOMO steps are presented in Algorithm 1, which can be summarized as: (i) all inactive nodes have their inputs changed randomly to another valid value; functions are changed with a probability \(p_{f}\); (ii) a random node (c) is chosen, in order to be mutated using SOMO; (iii) c has its function modified randomly with a probability \(p_{f}\); (iv) a random input (e) is chosen from c. Step (i) ensures that new genetic material is generated before performing the actual mutation. With the random input (e) chosen, SOMO identifies the best input considering all previous nodes in the genotype and then performs the mutation.

The identification of the most suitable node is based on semantics. Initially, it calculates the score of every node of the genotype that may be connected to the mutated node c. If more nodes receive the same score, the node closest to the program inputs is preferred. The score reflects the Hamming distance (HD). In the sequence, all left-sided nodes from the mutated node c are connected at e and simulated in three different ways: (a) using the function of the node being connected, (b) forcing e to logic zero (\(val^{ [0]}_{e=0}\)), and (c) forcing e to a logic one (\(val^{[0]}_{e=1}\)). The desired input value is denoted as req and it can be equal to ‘0’, ‘1’ or ‘X’, where ‘X’ means that it does not matter what Boolean value the input e takes. The value of req is determined by using the ternary operator \({ \Theta }\) and the reduction operator \(\bigodot \), defined in Equations 1 and 2. The term [o] in superscript points to a Boolean value associated with a program output node o.

In SOMO, the mutated input e of c is connected to the best node in the genotype of the current generation. Differently from the standard CGP, SOMO uses \(\lambda \) = 1 and a different population initialization. In SOMO, the population is started with a candidate solution having no active gate in order to maximize the efficiency and minimize the number of active gates of the evolved solutions [11].
4 Proposal and Methods
Here, we evaluate different approaches using SOMO aiming at discovering if using an appropriate optimization step helps it to obtain better GRNs. We propose the use of SOMO with SAM as (i) SOMO can obtain a feasible solution quickly but with many logic elements, and (ii) SAM is appropriate for reducing the number of logic elements. The proposal is presented in Algorithm 2.

In [11], the percentage of inactive nodes to be mutated (\(p_{q}\)) is equal to 100. Here, we analyze the performance of SOMO when using \(p_{q}\) = 50% with SAM as optimization step. In addition, as highlighted in [11], SOMO usually gets stuck in local optima. To avoid this issue, we use a restart strategy for exploiting different locations of the search space. The restart strategy consists of initializing a new population and restart the search for every k objective function evaluations.
The criteria for evaluating and comparing methods are varied, but [20] presented a set of benchmark problems when considering scRNA-Seq data for inferring GRNs. This set is composed of toy, curated, and real datasets. For toy and curated models, we have the ground-truth network, which facilitates the evaluation of new methods. Also, there is an extensive comparison between several algorithms from the literature using the pipeline provided in [20].
In this paper, we use curated scRNA-seq data in the form of pseudotime-series. These datasets were made considering 2,000 cells with three configurations: 0%, 50%, and 70% dropout rates. For each configuration, 10 datasets are given. Furthermore, each problem has a different number of pseudotimes.
As each pseudotime gives information about one possible cell trajectory, for each pseudotime one GRN is inferred. Then, the final GRN is given by merging the partial GRNs discovered for each pseudotime. This final GRN is evaluated considering the pipeline presented in [20] which considers the area under the precision-recall curve (AUPRC) and the area under the receiver operating characteristic curve (AUROC) values for comparison. AUPRC is calculated as the area under the precision-recall (PR) curve and shows the trade-off between precision and recall across different decision thresholds. The x-axis of a PR curve is the recall and the x-axis is the False-Positive Ratio (FPR). Considering TP as True Positive, FP as False Positive and FN as False Negative, we can define Precision = \(\frac{TP}{TP+FP}\) and Recall = \(\frac{TP}{TP+FN}\). For AUROC, the ROC curve is plotted considering the False-Positive Ratio (FPR) on the x-axis and the Recall, on the y-axis. Recall was defined previously and FPR = \(\frac{FP}{TN+FP}\). An excellent model has AUC close to 1, which means it has good separability. For many real-world datasets, particularly medical datasets, the fraction of positives is often less than 0.5, meaning that AUPRC has a lower baseline value than AUROC [6].
5 Computational Experiments
Computational experiments were conducted to analyze the performance of CGP mutation operators when applied to the inference of GRNs using the benchmark scRNA-Seq time-series data from [20]. Here, we highlight whether obtaining feasible solutions in a faster way has a positive impact on the quality of the solutions. Also, we analyze the importance of reducing the number of logic elements. The computational experiments are composed of two parts: (i) the performance of CGP variants are compared and, (ii) the results obtained by the two best CGP approaches are compared to those found by GENIE3.
The problems considered in our analysis are presented in Table 1, with information of the number of genes (#Genes) and the number of pseudotimes (#Pseudotimes). These problems are curated models with 2,000 cells and are presented in three different configurations considering 0%, 50%, and 70% dropout.
In order to remove or soften the effects of technical and biological variations as well as obtain a single curve that represents gene expression over time, we use data approximation via cubic smoothing splines implemented in Python and distributed through the library csapsFootnote 1. Then, the data is binarized with Bikmeans through Gene Expression Data Pre-Processing Tool (GEDPROTOOLS)Footnote 2. All methods were implemented in C++ and the source codes are availableFootnote 3.
The experiments were run in a Ubuntu Server 20.04 LTS (HVM) with 16 vCPUs Intel(R) Xeon(R) CPU E5-2666 v3 @ 2.90GHz and 30GB RAM and the results are evaluated using the pipeline presented in [20] which considers the AUPRC and AUROC values for comparison.
5.1 Comparative Analysis of the CGP Techniques
We compare the standard CGP with SAM and the CGP with SOMO in different approaches: (i) CGP: Standard CGP using SAM for obtaining the first feasible solution and for optimizing; (ii) SOMO: CGP using SOMO without optimization step; (iii) SOMO-SAM: CGP using SOMO with optimization step; (iv) SOMO-SAM-R: CGP using SOMO with optimization step and evolutionary search restart; (v) SOMO-SAM-PQ50: CGP using SOMO with optimization step and \(p_q\) = 50%. The suffix R means that the evolutionary process is restarted with a different initial population every 1,000 evaluations.
Here, we aim to evaluate the performance of SOMO with an appropriate optimization step. SOMO is able to obtain feasible solutions in a faster way than other approaches but this solution usually has many logic elements.
For the standard CGP, we use \(\lambda \) = 4 and random population initialization. When considering SOMO, we use \(\lambda \) = 2, \(p_{f} = 0\) and the population initialization suggested in [11]. Also, in [11] the value of \(n_c\) is variable, considering a multiple of the number of standard gates required to implement each circuit. However, the definition of the number of gates to implement a circuit cannot be easily defined a priori and, hence, we fixed \(n_c\) as usually considered when using the standard CGP and used only \(n_c = 100\). The other parameters are \(n_r = 1\) and \(lb=n_c\). For each problem, 5 independent runs were performed with a maximum of 100,000 objective function evaluations. For SOMO-SAM-R, the number of evaluations to restart is 1,000 and was defined during preliminary experiments, analyzing the average number of evaluations needed to find a feasible solution. Also, the accumulated number of evaluations is used in the stop criteria.
Tables 2 and 3 present the results of the CGP methods, respectively, for AUPRC and AUROC. The best (maximum), first quantile (Q1), Median, Mean, third quantile (Q3), and standard deviation values of the results are shown, and the best values are highlighted in boldface. Also, the methods with an asterisk have statistical difference, when considering Dunn’s test for 95% of confidence.
Based on these results, one can see that the results of SOMO-SAM and SOMO-SAM-R are equal in some cases. This is expected as the first feasible solution was obtained before reaching the first restart point (1,000 evaluations). In general, most CGP approaches have no statistical difference when compared to each other, both in AUPRC and AUROC. When considering the median, we count the times that each algorithm reached the best result. Values in parentheses consider the statistical tests and when there is no statistical difference, all methods score. This counting is shown in Table 4.

Results for the problems considering all scenarios.

PPs considering AUPRC and AUROC for the best approaches.
Also, the approaches using an optimization step performed better than the other ones. The restart scheme helps to escape from local optima but it does not provide good results. Furthermore, the results when using \(p_{q}\) = 50% are better than the standard values presented in [11]. The SOMO population initialization is essential for a good performance of the algorithm as well as using \(\lambda \) = 1.
Finally, based on Table 4, one can conclude that the best methods are SOMO-SAM and SOMO-SAM-PQ50 when AUPRC is considered. For AUROC, the best algorithms are SOMO-SAM and SOMO. However, here we consider one approach using the standard SOMO with SAM as optimization step (SOMO-SAM) and another approach varying the parameter \(p_{q}\), namely SOMO-SAM-PQ50.
5.2 Comparative Analysis with GENIE3
As observed in Sect. 5.1, the two best CGP variants are SOMO-SAM and SOMO-SAMPQ50, and we compare them here with GENIE3. According to [20], GENIE3 is the best algorithm for inferring GRNs. Performance profiles (PPs) [5] were used in order to analyze the relative performance of the algorithms. Considering p as a particular problem (model) and s as a particular solver, \(\rho (p,s)\) is defined as the performance ratio within a factor of \(\tau \) of the best possible ratio.
From PPs, it is possible to extract: (i) the approach that obtained the best results for most problems (largest \(\rho (1)\)), (ii) the most reliable approach (smaller \(\tau \) such that \(\rho (\tau ) = 1\)), and (iii) the best overall performance (largest area under the performance profiles curves). Moreover, boxplots of the results are presented in Fig. 2. Also, Kruskal Wallis statistical test and Dunn’s post hoc test were carried out and the results show that there is statistical difference only when comparing CGP approaches with GENIE3.
Based on the PPs presented in Fig. 3 is possible to conclude that: (i) GENIE3 has the best performance in most of the problems (largest \(\rho (1)\)), followed by SOMO-SAM and SOMO-SAM-PQ50, respectively. (ii) GENIE3 is the most reliable variant (smallest \(\tau \) such that \(\rho (\tau )=1\)), followed by SOMO-SAM-PQ50 and SOMO-SAM, respectively, and; (iii) GENIE3 presents the best overall performance (largest AUC), followed by SOMO-SAM-PQ50 and SOMO-SAM, respectively. Thus, GENIE3 is a good choice when considering only AUPRC. However, as highlighted in Sect. 4, for many real-world medical datasets the fraction of positives is often less than 0.5.
On the other hand, when considering the performance profiles of AUROC, we can conclude that: (i) GENIE3 hast the best performance in most of the problems, followed by SOMO-SAM and SOMO-SAM-50, respectively; (ii) SOMO-SAM and SOMO-SAM-PQ50 are the most reliable variants, and; (iii) SOMO-SAM presents the best overall performance, followed by SOMO-SAM-PQ50 and GENIE3, respectively. Then, SOMO-SAM highlights the importance of using an appropriate optimization step.
Also, SOMO is very sensitive to the parameter \(p_{q}\), i.e. keeping some inactive nodes unchanged before performing SOMO helps to obtain better results. One possible reason is the fact that, when using SAM, we adopt \(\lambda = 4\), then, CGP is able to create more diverse offspring than with \(\lambda = 1\) as the standard SOMO uses. However, only when considering mCAD, approaches that use CGP obtained better results on AUPRC and AUROC. With respect to mCAD it is interesting to highlight that this problem is the only one with 2 pseudotimes. The evaluation pipeline considers the “top k” regulatory relationships and, when merging the 2 solutions to obtain the final GRN, it is possible to construct a smaller GRN (less number of regulatory relationships) that consider the most important regulatory relationships leading to a better evaluation.
The statistical tests show that there is no statistical difference between the CGP approaches. The statistical difference is observed only when comparing the CGP variants with GENIE3, reinforcing the superiority of GENIE3 as shown in the AUPRC boxplots.
6 Conclusions and Future Work
Here, we analyze the performance of CGP mutation operators applied when inferring GRN using benchmark curated scRNA-seq time series data. These approaches include mainly two aspects: (i) a proposal using SAM to optimize the solution obtained by SOMO, and (ii) a parameter sensitivity analysis for SOMO.
We compared the CGP approaches, and the best-obtained CGP methods with GENIE3. The results show that modifying \(p_{q}\) to 50% helped CGP to obtain better results, as a more diverse offspring is generated. GENIE3 performed better in most problems, except in mCAD. The mCAD problem has only two pseudotimes and the CGP solutions’ merge can obtain a smaller GRN that is better evaluated in the pipeline. Also, there is no statistical difference between the CGP approaches, but there is when comparing the CGP variants with GENIE3. According to PPs, GENIE3 obtained the best results in general but one can highlight that both SOMO-SAM and SOMO-SAM-PQ50 are more reliable when AUROC is considered. Furthermore, the use of SOMO for obtaining faster first feasible solutions and SAM as an appropriate optimization step helps CGP to obtain better results when inferring gene regulatory networks when compared to the standard SOMO and to the standard CGP. Thus, one can conclude that using SAM with SOMO improves the inferred GRNs.
As future work, we intend to explore even more the SOMO parameters, considering other values of \(p_{q}\) and \(p_{f}\) in the context of the inference of GRNs and the impact of \(n_{c}\) when adopting values of \(\lambda > 1\). Also, we intend to investigate the impact of the number of pseudotimes in the merging step of CGP.
References
Aalto, A., Viitasaari, L., Ilmonen, P., Mombaerts, L., Gonçalves, J.: Gene regulatory network inference from sparsely sampled noisy data. Nat. Commun. 11(1), 1–9 (2020)
Banf, M., Rhee, S.Y.: Computational inference of gene regulatory networks: approaches, limitations and opportunities. Biochimica et Biophysica Acta (BBA) Gene Regul. Mech. 1860(1), 41–52 (2017)
Chan, T.E., Stumpf, M.P., Babtie, A.C.: Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 5(3), 251–267 (2017)
Chen, S., Mar, J.C.: Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinf. 19(1), 1–21 (2018)
Dolan, E.D., Moré, J.J.: Benchmarking optimization software with performance profiles. Math. Programm. 91(2), 201–213 (2002)
Draelos, R.: Measuring performance: Auprc and average precision (2019). glassboxmedicine.com/2019/03/02/measuring-performance-auprc/
Friedman, J.H.: Greedy function approximation: a gradient boosting machine. Ann. Statist., pp. 1189–1232 (2001)
Gebert, J., Radde, N., Weber, G.W.: Modeling gene regulatory networks with piecewise linear differential equations. Eur. J. Oper. Res. 181(3), 1148–1165 (2007)
Goldman, B.W., Punch, W.F.: Reducing wasted evaluations in cartesian genetic programming. In: Krawiec, K., et al. (eds.) EuroGP 2013. LNCS, vol. 7831, pp. 61–72. Springer, Heidelberg (2013). https://doi.org/10.1007/978-3-642-37207-0_6
Haghverdi, L., Büttner, M., Wolf, F.A., Buettner, F., Theis, F.J.: Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13(10), 845 (2016)
Hodan, D., Mrazek, V., Vasicek, Z.: Semantically-oriented mutation operator in cartesian genetic programming for evolutionary circuit design. In: Proceedings of the 2020 Genetic and Evolutionary Computation Conference, pp. 940–948 (2020)
Irrthum, A., Wehenkel, L., Geurts, P., et al.: Inferring regulatory networks from expression data using tree-based methods. PloS One 5(9), e12776 (2010)
Jackson, C.A., Castro, D.M., Saldi, G.A., Bonneau, R., Gresham, D.: Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. Elife 9, e51254 (2020)
Liu, S., Trapnell, C.: Single-cell transcriptome sequencing: recent advances and remaining challenges. F1000Research 5 (2016)
Ma, B., Jiao, X., Meng, F., Xu, F., Geng, Y., Gao, R., Wang, W., Sun, Y.: Identification of gene regulatory networks by integrating genetic programming with particle filtering. IEEE Access 7, 113760–113770 (2019)
McCall, M.N.: Estimation of gene regulatory networks. Postdoc J. Postdoc. Res. Postdoc. Affairs, 1(1), 60 (2013)
Miller, J.F.: Cartesian genetic programming. CGP, pp. 17–34 (2011)
Miller, J.F., Thomson, P., Fogarty, T.: Designing electronic circuits using evolutionary algorithms. arithmetic circuits: A case study (1997)
Moerman, T., et al.: GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35(12), 2159–2161 (2019)
Pratapa, A., Jalihal, A.P., Law, J.N., Bharadwaj, A., Murali, T.: Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. methods 17(2), 147–154 (2020)
Qiu, X., Mao, Q., Tang, Y., Wang, L., Chawla, R., Pliner, H.A., Trapnell, C.: Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14(10), 979 (2017)
Huynh-Thu, V.A., Sanguinetti, G.: Gene regulatory network inference: an introductory survey. In: Sanguinetti, G., Huynh-Thu, V.A. (eds.) Gene Regulatory Networks. MMB, vol. 1883, pp. 1–23. Springer, New York (2019). https://doi.org/10.1007/978-1-4939-8882-2_1
Setty, M., et al.: Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotech. 34(6), 637–645 (2016)
da Silva, J.E.H., et al.: Inferring gene regulatory network models from time-series data using metaheuristics. In: IEEE Congress on Evolutionary Computer (CEC), pp. 1–8. IEEE (2020)
da Silva, J.E.H., Bernardino, H.S., de Oliveira, I.L.: Inference of gene regulatory networks from single-cell RNA-sequencing data using cartesian genetic programming (under review). In: Bioinformatics, pp. 1–8. Oxford (2021)
Streichert, F., et al.: Comparing genetic programming and evolution strategies on inferring gene regulatory networks. In: Deb, K. (ed.) GECCO 2004. LNCS, vol. 3102, pp. 471–480. Springer, Heidelberg (2004). https://doi.org/10.1007/978-3-540-24854-5_47
Trapnell, C., et al.: The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32(4), 381 (2014)
Wang, R.S., Saadatpour, A., Albert, R.: Boolean modeling in systems biology: an overview of methodology and applications. Phys. Biol. 9(5), 055001 (2012)
Acknowledgements
We thank the support provided by FAPERJ, FAPESP, FAPEMIG, CAPES, CNPq, UFJF, and Amazon AWS.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Silva, J.E.H.d., Bernardino, H.S., de Oliveira, I.L., Vieira, A.B., Barbosa, H.J.C. (2021). On the Analysis of CGP Mutation Operators When Inferring Gene Regulatory Networks Using ScRNA-Seq Time Series Data. In: Britto, A., Valdivia Delgado, K. (eds) Intelligent Systems. BRACIS 2021. Lecture Notes in Computer Science(), vol 13073. Springer, Cham. https://doi.org/10.1007/978-3-030-91702-9_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-91702-9_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91701-2
Online ISBN: 978-3-030-91702-9
eBook Packages: Computer ScienceComputer Science (R0)