
Abstract
MicroRNAs (miRNAs) are crucial regulators of gene expression, including in diseases such as cancer. Although machine learning methods have shown promise in predicting miRNA-target interactions, they encounter challenges related to imbalanced classes and false positives. To tackle these issues, this study proposes a GNN-based model, using a variant of GraphSAGE algorithm named HinSAGE, which integrates validated miRNA-mRNA and mRNA-mRNA interactions with cancer-related gene expression data. Results show that our approach effectively learns miRNA-target interaction patterns from the graph structure and node features. The model achieves 77% precision, 80% recall, 78% F1-score, and 86% ROC AUC on the test data. It competes well with related approaches, reaching an F1-score of approximately 90% on a common test set. Thus, GNNs offer a promising avenue for studying miRNA-target interactions, providing balanced predictive power and improved precision through negative interaction sampling from the graph.
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001, and by grants from FAPERGS [21/2551-0002052-0] and CNPq [308075/2021-8].
Access provided by University of Notre Dame Hesburgh Library. Download conference paper PDF
Similar content being viewed by others


1 Introduction
MicroRNAs (miRNAs) are small non-coding ribonucleic acid (RNA) molecules that regulate gene expression in post-transcriptional stages, inhibiting the expression of specific genes by messenger RNA (mRNA) silencing. Recent studies indicate that a single miRNA can target many different mRNAs, while a particular mRNA can be regulated by a set of miRNAs, simultaneously or in a context-dependent manner. Moreover, abnormal miRNA expression has been associated with the development and progression of different human diseases, including those related to the endocrine system and cancer [10]. Thus, miRNAs are important actors in the complex regulatory network governing gene expression in physiological and pathological processes.
Unraveling miRNA-mRNA target interactions is a fundamental step for discovering the regulatory network governed by miRNAs and their functional role within an organism. Due to the numerous potential target sites for a given miRNA, experimentally confirming every possible target candidate is impractical both in terms of cost and time. Machine learning (ML) has been increasingly explored to predict candidate targets using a series of descriptors (i.e., features) of the miRNA-target mRNA interaction defined from experimentally characterized interactions and prior knowledge [13], filtering possible miRNA targets.
However, miRNA target prediction is still considered an open problem due to several challenges. First, manually engineered features based on biological assumptions capture only partially the numerous characteristics that influence the effectiveness of an interaction between miRNA and mRNA target [13]. The contribution of single features to the degree of repression of the miRNA target is still hard to completely determine. Second, the multiplicity of miRNA-target interaction patterns, often in a context-dependent manner, introduces large complexity into the problem. Third, there is an inherent class imbalance issue, as the majority of validated data refers to functional miRNA targets, i.e., positive examples. This generates a bias towards large false positive predictions [11].
Over the past years, numerous deep learning (DL) methods have been developed, with various successful applications in biology and medicine. Among these, we highlight the DL algorithms in graphs, known as Graph Neural Networks (GNNs), and their capacity for solving complex problems where data requires a representation in non-Euclidean domains [1, 16]. GNNs have found successful applications in identifying associations between miRNAs and diseases (e.g., [4]); however, they are still little explored in the computational prediction of miRNA targets. Thus, in this work we aim to answer the question: can GNNs precisely predict miRNA targets from known miRNA-mRNA target interactions and biological evidence regarding miRNAs and mRNAs expression?
We propose an approach based on the HinSAGE algorithm and heterogeneous graphs containing prior knowledge about validated miRNA-mRNA target and mRNA-mRNA interactions. HinSAGE is a derivative of the GraphSAGE algorithm [3], an stochastic generalization of graph convolutions that can leverage node feature to efficiently generate embeddings on unseen data. Moreover, instead of using manually engineered features from known miRNA-target interactions, we use gene expression profiles as node features. Gene expression is the final product in the process of gene regulation, which is affected by molecular factors that are usually used as predictive features individually or combined, such as sequence complementarity, sequence conservation, favorable thermodynamics, and accessibility of the miRNA binding site. Here, we focus on miRNA-target interactions associated with cancer, motivated by the role of dysregulated miRNAs in cancer pathophysiology and as potential biomarkers [10].
Our main contributions are twofold. First, we propose and evaluate a prediction approach based on the GraphSAGE algorithm that integrates an graph and expression profiles to predict miRNA-target interactions. To the best of our knowlegde, this is the first work to explore GNNs for miRNA-target prediction in human cancer. Second, we conduct a detailed investigation of several hyperparameters involved in the algorithm and decisions regarding dataset construction, evaluating their impact on the addressed prediction problem.
2 Related Works
Several tools have been developed for miRNA target prediction, with ML and DL being recurrent among solutions. Since the related literature is vast [13], here we review the most relevant works for the proposed approach. TargetScan [9] is a widely used tool that employs linear regression and ML techniques for miRNA target prediction. However, it has limited sensitivity for targets without good evolutionary conservation. PITA [7] considers the accessibility of the binding site on the target, evaluating the scores for microRNA-target interactions by using energy-based secondary structure prediction algorithms. Both methods rely on specialist-designed features.
More recent approaches include miRAW [12], DeepTarget [8], and DeepMirTar [15], which are based on DL and aim to learn better representations directly from raw data, eliminating the need for manual feature extraction. They implement autoencoders and adopt distinct approaches for defining negative samples, such as generating mock examples or building negative examples based on experimentally verified data. We note that these approaches do not explore the graph structure and some of the works lack evaluation on independent test datasets.
Finally, we highlight comprehensive databases containing computationally predicted interactions: TarBase v8 [6] and mirDIP [14]. These databases integrate data from various sources and provide confidence scores for their predictions. We note that a main challenge faced was the difficulty of identifying, up to the time of writing this paper, a model for miRNA target prediction based on the concept of GNNs. It should be noted that GNNs have been previously applied in prediction tasks related to miRNAs, but with a special focus on predicting the association of miRNAs with human diseases (e.g., [4]) - a task that has different objectives from the problem modeled in this study. Moreover, a previous study has explored GNNs in the miRNA-target prediction problem, although focusing on data from Camellia sinensis and without integrating gene expression profiles into their framework [2].
3 Methodology
GNNs are DL algorithms designed to operate on graph-structured data. The learning of node representations in the graph, i.e., the generation of embeddings, is achieved through iterative updates that aggregate the representations of neighboring nodes and the node itself from previous iterations. This information can be used for tasks such as link prediction (our interest) or node classification. In this section, we detail how we collected and integrated the interactions to define the graph structure and the gene expression data used as node features in the model training process, as depicted in Fig. 1-a. We also explain the proposed learning approach based on the HinSAGE algorithm (Fig. 1-b).

Summary of the approach proposed. a) The graph consists of miRNAs (red node) and mRNAs (pink nodes) with their features vectors, and the known interactions among them (solid lines). b) The inductive learning process by the HinSAGE algorithm considering potential miRNA-mRNA interactions (dashed lines) using sampled 2-hop neighbors. (Color figure online)
3.1 Data Collection, Preprocessing and Integration
miRNA-mRNA and mRNA-mRNA Interaction Data. We obtained interactions from RNAInter v4.0 [5], a comprehensive database that integrates RNA interactome data, including experimentally validated and computationally predicted interactions. The current version has over 47 million annotated interactions for 156 species. RNAInter categorizes the type of experimental evidence for each interaction as strong or weak, and also calculates a confidence score per interaction based on characteristics such as experimental evidence confidence, scientific community confidence given by the number of article citations, and the number of different tissues or cells in which the interaction was identified – all integrated using a sigmoid function. We downloaded miRNA-mRNA and mRNA-mRNA interactions related to humans, removing those with inconsistent values, such as undefined miRNA or mRNA identifiers, and those derived from computational prediction to improve the quality of the training data.
Gene Expression Data in Cancer. Gene expression data for miRNAs and mRNAs was obtained from The Cancer Genome Atlas (TCGA) project, one of the leading consortia in cancer genomics. The data is publicly accessible through the GDAC FireBrowse portalFootnote 1. We downloaded gene expression datasets for primary solid tumor samples (PT) and normal tissue (NT) samples (i.e., non-tumor) preprocessed following well-established bioinformatics protocols. We only considered those cancers that did not exhibit significant imbalance between the number of tumor and normal tissue samples for miRNAs and mRNAs expression, resulting in 15 tumor types (see Supplementary MaterialFootnote 2). The data was transformed to the log2 scale and a measure of differential expression, the log fold change (logFC), was calculated for each miRNA and mRNA in relation to each tumor type. The logFC expresses the ratio between the mean gene expression in PT vs. NT samples for a specific miRNA or mRNA, in a specific cancer type. A higher absolute value of logFC is assumed to indicate greater biological relevance in the studied domain. The step of summarizing expression patterns through logFC values generates a feature vector of length 15 for each miRNA and mRNA in the graph.
Data Integration for Graph Generation. We integrated the interaction data obtained from the RNAInter database with the gene expression data collected from FireBrowse using the unique identifier available for each miRNA and mRNA in the collected database. During this step, miRNA-target interactions without any identified expression records were also removed. This removal aimed to mitigate the induction of inconsistent results during the subsequent training of the proposed model. The final interaction network comprises over 100,000 miRNA-target interactions and nearly 50,000 mRNA-mRNA interactions. The network has a total of 2,617 miRNA nodes and 17,252 mRNA nodes. The number of interactions according to interaction type, evidence type, and score threshold is shown in Fig. 2.

Summary of collected miRNA-mRNA and mRNA-mRNA interactions.
3.2 Model Training and Evaluation
In this work, we are interested in conducting a link prediction task handling a large amount of data, integrating node features into the learning process, and working with a heterogeneous graph. Based on the literature [16], we chose a variant of GraphSAGE [3] extended for heterogeneous graphs, named HinSAGEFootnote 3. The algorithm generates embeddings by sampling and aggregating features from the local neighborhood of a node, instead of training individual embeddings focused solely on the node. The number of observed neighbors can be parameterized, both in terms of the number of hops (K) and the number of neighboring nodes to be sampled at each hop. This behavior can be observed in Fig. 1, where the value \(K=2\) represents the number of hops (i.e., depth of search or number of HinSAGE layers). The neighborhood aggregation strategy is applied such that neighbors are randomly selected and fused together by edge type.
The predictive models were developed using the HinSAGE implementation provided by the StellarGraph library. The base graph was built as explained in Sect. 3.1. The hyperparameters were initially defined as follows: batch size equal to 200; 300 epochs of training; learning rate of 0.001 with Adam optimizer; two hops (\(K=2\)); number of nodes sampled per hop equal to [8,4]; HinSAGE layer sizes setup as [32,32]; binary Cross-Entropy cost function; ReLU activation function; feature update rule based on the Graph Convolutional Network (GCN) aggregator. Variations in the hyperparameter values were tested and evaluated in a series of experiments that will be detailed later.
Our experiments were based on ten random divisions of the data into training, validation, and test sets. Considering our inductive domain, the training, validation, and test sets are subgraphs derived from reductions of the original graph, aiming to generate independent data for the different stages involved in model development (Table 1). We used the method EdgeSplitter provided by StellarGraph, which takes as input both the graph to be subdivided and a percentage p relative to the total number of edges in the provided graph, determining the number of positive and negative edge samples to be sampled from it. Positive edges are sampled from the actual set of edges present in the graph to maintain graph connectivity. Negative edges are randomly created by sampling pairs of miRNA-mRNA that are not connected by an interaction in the original graph. This sampling of negative examples aims to improve the model’s ability to identify false positive edges, by mitigating class imbalance. The value of the hyperparameter p was adjust according to the set of interactions included in the creation of the graph, since it needs to be compatible with the number of negative interactions that can be randomly generated from the input graph. Model performance evaluation was based on the accuracy, precision, recall, F1-Score, and ROC AUC metrics.
4 Experiments
Two main sets of experiments were defined for the development of this study.
The first set, S1, is focused on analyzing the potential of the HinSAGE algorithm for miRNA target prediction using a graph-based learning approach. We evaluate the influence of aspects related to the construction of the interaction dataset and the configuration of the algorithms’s hyperparameters on the results achieved by the HinSAGE model. We define 20 experimental scenarios organized into seven groups (Table 2), each of which with a specific experimental goal: G1: evaluation of the impact of using different datasets to define the base graph used in the model training, constructed by removing mRNA-mRNA interactions and with different score filters; G2: evaluation of the impact of removing mRNA-mRNA interactions from the base graph; G3: evaluation of the impact of reducing the number of training epochs; G4: evaluation of the impact of varying the batch size; G5: evaluation of the impact of varying the number of sampled neighboring nodes; G6: evaluation of the impact of varying the size of the hidden layers; G7: evaluation of the impact of varying the learning rate. Given the high computational cost involved in running each experiment, at the end of each experimental group, we identified the most promising configuration to be employed in the next group. Thus, best hyperparameter values found in previous experiments are carried over in the next group of experiments.
The second set, S2, aims to compare the approach developed in this study (based on the best model generated in the set S1) with other methods proposed in the literature. Due to lack of similar approaches, it was not possible to perform an equivalent comparison based on end-to-end graph analysis using GNNs. We conducted the comparison by focusing on the results of the models’ predictions for the test data, considering miRNA-target interactions found in common between the test set created in this study and the test sets used in the related works. We used the predictions provided by the authors of the original papers. To avoid greatly restricting the size of the common dataset due to the intersection of multiple methods, our comparison was performed pairwise with each selected related work. Since most of related works do not provide predicted probabilities, we did not use the ROC AUC metric in this analysis.
5 Results and Discussion
The next sections present and discuss the results for the two sets of experiments. We note that due to space constraints, we focus on the most meaningful findings. The complete results are provided as supplementary materialFootnote 4.
5.1 S1: Exploring the Predictive Performance of HinSAGE
In the experiments of G1, we aimed to gain a better understanding of the model’s behavior with different datasets used for training. We compared the original dataset, including both miRNA-mRNA and mRNA-mRNA interactions (Experiment 00), with a version excluding mRNA-mRNA interactions (Experiment 01) using the default hiperparameters values (Sect. 3.2). The results obtained during training showed promising numbers, with an accuracy close to 75% for both cases. However, upon examining the accuracy and loss throughout the training epochs, we identified characteristics of overfitting occurring from epoch 50 onwards. Moreover, we did not observe any significant performance change by removing mRNA-mRNA interactions, thus, we maintained them.

Performance evaluation for selected experiments of G1 in terms of mean and standard deviation for 10 random training, validation and test sets.
We also evaluated the influence of various filters applied to the confidence score associated to each interaction. Experiments 02 to 05 filtered interactions based on thresholds of 0.2, 0.3, 0.4, and 0.5 (Table 2), thus reducing the number of interactions in the graph (Table 1). We noticed a sequence of performance improvements for both training and testing data at the end of the 300 training epochs. The initial filter (score \(>=\) 0.2 in Experiment 02) already showed improvements compared to Experiment 00. However, we also observed a highly negative overfitting effect in some cases, such as in Experiment 02 and, particularly, in Experiment 05. We highlight the results of Experiment 04, which proved to be the most promising in group G1, demonstrating an apparent improvement without a significant tendency towards overfitting. Figure 3 summarizes the training, validation, and test results for the main experiments of G1. It can be concluded in advance that applying the score filter does indeed influence the achieved results, and reducing the number of interactions described as strong (which decrease for larger thresholds) hinders the prediction of interactions, resulting in significant dispersion in the obtained results.
Next, in G2, we aimed to analyze whether the use of mRNA-mRNA interactions could influence the results obtained with filtered datasets. Therefore, Experiments 06 and 07 correspond to Experiments 04 and 05, respectively, but considering only miRNA-mRNA interactions. Once again, removing these interactions influenced the results, but the impact was not consistent between both experiments. We noticed that removing mRNA-mRNA interactions introduced overfitting in the experiment (Experiments 06 vs 04), whereas when comparing Experiments 07 vs 05, the training became more stable and showed less overfitting after removing these interactions, but without noticeable increase in the predictive performance. Hence, Experiment 04 (G1) remained the best scenario.

Performance evaluation for G3 in terms of mean and standard deviation for 10 random training, validation and test sets.
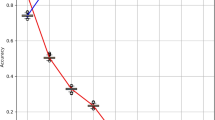
Since we observed some signs of overfitting, in G3 we evaluated the reduction in the number of training epochs to mitigate overfitting and reduce the chances of predicting false positive interactions. Results are shown in Fig. 4 and they can be compared to Experiment 04, as the other configurations remain the same between the two experiments. We can observe that the training curves for 100 epochs exhibit more appropriate behavior, with close training and validation performances. This means that the confidence in the generalization power of the generated model becomes stronger. Moreover, the performances in the test set were quite satisfactory, although there was a slight reduction in values compared to the performance on the test data for the model generated in Experiment 04. The precision varied mostly with values above 75% and the ROC AUC had a median value close to 85%. The following experiments will adopt 100 epochs.
Experiments of G4 aimed to initiate the investigation of the hyperparameter values involved in the HinSAGE algorithm by exploring variations in the batch size, which was initially set to 200. Four distinct values were tested: 1 (Experiment 09), 50 (Experiment 10), 100 (Experiment 11), and 300 (Experiment 12). A batch size of 1 showed a training behavior described as stochastic, presenting overfitting within the first ten epochs of training. This characteristic was also present with a batch size of 50, which showed a large standard deviation for the first 25 epochs. Experiments 11 and 12, obtained more appropriate results. The latter, highlighted in Fig. 5, had a slight performance improvement when compared to Experiment 08, particularly in terms of performance variation across the 10 executions. The following experiments will apply a batch size of 300.

Performance evaluation for selected experiment in G4 in terms of mean and standard deviation for 10 random training, validation and test sets.
In G5, we investigated the impact of varying the number of sampled neighbor nodes in the HinSAGE algorithm on the generation of embeddings (i.e., num_sample). We kept the number of hops fixed at \(K=2\) and tested two variations: [6,3] (Experiment 13) and [12,6] (Experiment 14). We observed that reducing the number of sampled neighbors (Experiment 13) resulted in increased performance dispersion between training and validation, while increasing the number of sampled neighbors (Experiment 14) led to a decrease in the dispersion of these performances. Moreover, Experiment 14 (Fig. 6) also showed improved performance compared to Experiment 12. The medians for all metrics were approximately 80% or higher. A more balanced performance between recall and precision was observed, resulting in an increase in the F1-Score metric. The ROC AUC metric had values very close to 90%. The better performances in the training, validation, and test datasets motivated the selection of Experiment 14 as the best scenario so far.

Performance evaluation for selected experiment in G5 in terms of mean and standard deviation for 10 random training, validation and test sets.

ROC curves for the test sets in Experiment 14.
Finally, experiments in G6 and G7 aimed to compare variations in the size of hidden layers and in the learning rate, respectively. Our results indicate that increasing the dimensions of the hidden layers (Experiments 15 and 16) led to increased overfitting and had no positive effect on the maximum performance obtained by the model. On the other hand, reducing the size of the hidden layers (Experiment 17) caused the model to loose predictive power compared to the other evaluated scenarios, but mainly Experiment 14. Regarding the analysis for the learning rate, when its value was increased to 0.01 and 0.005, we observed more signs of overfitting without effective improvement in the achieved predictive performance. Conversely, decreasing the learning rate to 0.0001 had a detrimental effect on the predictive model’s performance, causing underfitting of the model. In this case, the learning progress was very slow and the model failed to converge adequately for the fixed number of epochs (100).
Among all the evaluated scenarios in the set of experiments S1 (Table 2), the best model was the one originated from Experiment 14 and it will be further employed in the experiment set S2. Figure 7 shows the ROC curves and corresponding ROC AUC values for the test data considering the 10 runs of the 3-way holdout. The model’s performance appears to be quite stable, with ROC AUC values ranging from 86% to 88%. Additionally, the curve demonstrates a suitable growth, with a steeper increase in TPR (y-axis) compared to FPR (x-axis). For example, a TPR of 80% yields an FPR close to 20%.
5.2 S2: Comparison of HinSAGE Model with Related Works
The related works for which a direct comparison was possible with our test data were miRAW [12], TargetScan [9], and PITA [7]. We also used computationally predicted interactions from TarBase v8 [6] and mirDIP [14]. To conduct the comparison, we intersected our test data with the pre-computed target prediction results provided by each tool or method. The prediction datasets were obtained from the official platforms of the respective works (accessed on January 10, 2023).
Some preprocessing steps were required in some cases. For TargetScan, predictions are associated to a context++ score and targets with lower context++ scores are considered more representative. However, there is no clearly defined cutoff point to determine whether a given miRNA-mRNA interaction should be classified as positive or negative. Therefore, we decided to apply three distinct thresholds: –0.5, –0.3, and –0.2. Interactions with context++ scores lower than the applied threshold were classified as positive. For TarBase, we only used interactions related to humans and classified as ‘positive’ by the tool, indicating that they represent functional miRNA-mRNA interactions. For mirDIP, each interaction has a score assigned and described as: Very High, High, Medium, and Low. Very High indicates a high probability of the miRNA-mRNA interaction, while the probability decreases towards the Low designation. Due to the large volume of data, we obtained only the dataset with a score value described as Very High, inferring all resulting interactions as positives.
The results for this analysis are shown in Table 3. For all the related works with which a direct comparison was possible for the test instances, our proposed model based on the HinSAGE algorithm achieved results that are very close to or outperform other approaches. Excluding TarBase and mirDIP, which are databases rather than ML-based prediction methods, our work presents highly promising results. In almost all the performed comparisons, our model achieved an F1-score above 90%, indicating that the model developed in this work can predict positive and negative interactions with a high degree of confidence.
However, it should be noted that none of the related works focus on identifying miRNA-mRNA interactions associated with cancer. This limitation exists in comparing our results with the literature, given the difficulty of finding representative datasets for this context. Other critical challenges faced for this comparison were (i) the wide variety of predictive features used by previous works based on ML, turning the creation of the training dataset a slow and arduous process and (ii) the difficulty in running methods that are not made available through online tools, requiring local installation of the application and its dependencies.
6 Conclusion
The premise that guided the present work is that the development of a computational strategy based on methods capable of learning miRNA regulation patterns from the analysis of heterogeneous interaction networks (i.e., HinSAGE) could provide more robust predictions to noise and deficiencies in training data, as well as better capture the complexity involved in miRNA function. The use of graph-based DL algorithms not only introduces the advantage of exploring the graph structure in a much more comprehensive and robust way to discover patterns in miRNA-mRNA interactions, but also enables efficient handling of the data imbalance problem prevalent in this domain. Our experiments suggest that our approach was effective, reaching competitive performance and controlling false positive rates. Although there is no known equivalent work at the moment that can serve as a baseline for comparison to our work, the comparisons performed with computationally predicted interactions from other ML-based methods or deposited in databases have shown that the approach based on heterogeneous graphs and the application of the HinSAGE algorithm are highly promising. The performance achieved by our model stood out in various scenarios and demonstrated a suitable balance between recall and precision. Therefore, we believe that the objective of exploring the potential of the HinSAGE algorithm for miRNA target prediction in cancer has been successful, and the results obtained in this study serve as motivation for further exploration of this approach. Among possible directions for future work, we outline the application to different human diseases, the inclusion of other types of interactions in the heterogeneous networks, and the need to expand the comparison with other ML/DL algorithms or bioinformatics tools for miRNA-target prediction.
References
Cai, H., Zheng, V.W., Chang, K.C.C.: A comprehensive survey of graph embedding: problems, techniques, and applications. IEEE Trans. Knowl. Data Eng. 30, 1616–1637 (2018)
Feng, H., Xiang, Y., Wang, X., Xue, W., Yue, Z.: Mtagcn: predicting mirna-target associations in camellia sinensis var. assamica through graph convolution neural network. BMC Bioinf. 23(1), 1–18 (2022)
Hamilton, W., Ying, Z., Leskovec, J.: Inductive representation learning on large graphs. In: Advances in Neural Information Processing Systems, pp. 1024–1034 (2017)
Ji, C., Wang, Y., Ni, J., Zheng, C., Su, Y.: Predicting miRNA-disease associations based on heterogeneous graph attention networks. Front. Genet. 12, 727744 (2021)
Kang, J., et al.: RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 50, D326–D332 (2022)
Karagkouni, D., et al.: DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 46(D1), D239–D245 (2018)
Kertesz, M., Iovino, N., Unnerstall, U., Gaul, U., Segal, E.: The role of site accessibility in microRNA target recognition. Nat. Genet. 39(10), 1278–1284 (2007)
Lee, B., Baek, J., Park, S., Yoon, S.: deeptarget: end-to-end learning framework for microrna target prediction using deep recurrent neural networks. In: Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, pp. 434–442 (2016)
Lewis, B., Shih, I.H., Jones-Rhoades, M., Bartel, D., Burge, C.: Prediction of mammalian MicroRNA targets. Cell 115, 787–798 (2004)
Peng, Y., Croce, C.M.: The role of MicroRNAs in human cancer. Signal Transd. Target. Tpherapy 1(1), 1–9 (2016)
Pinzón, N., et al.: microRNA target prediction programs predict many false positives. Genome Res. 27(2), 234–245 (2017)
Pla, A., Zhong, X., Rayner, S.: miRAW: a deep learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts. PLOS Comput. Biol. 14, e1006185 (2018)
Schäfer, M., Ciaudo, C.: Prediction of the miRNA interactome - established methods and upcoming perspectives. Comput. Struct. Biotechnol. J. 18, 548–557 (2020)
Tokár, T., et al.: MirDIP 4.1 - Integrative database of human microRNA target predictions. Nucleic Acids Res. 46, D360–D370 (2017)
Wen, M., Cong, P., Zhang, Z., Lu, H., Li, T.: Deepmirtar: a deep-learning approach for predicting human mirna targets. Bioinformatics 34(22), 3781–3787 (2018)
Zhou, J., et al.: Graph neural networks: a review of methods and applications. AI Open 1, 57–81 (2020)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Vianna Fabiano, E.A., Recamonde-Mendoza, M. (2023). Prediction of Cancer-Related miRNA Targets Using an Integrative Heterogeneous Graph Neural Network-Based Method. In: Naldi, M.C., Bianchi, R.A.C. (eds) Intelligent Systems. BRACIS 2023. Lecture Notes in Computer Science(), vol 14197. Springer, Cham. https://doi.org/10.1007/978-3-031-45392-2_23
Download citation
DOI: https://doi.org/10.1007/978-3-031-45392-2_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-45391-5
Online ISBN: 978-3-031-45392-2
eBook Packages: Computer ScienceComputer Science (R0)